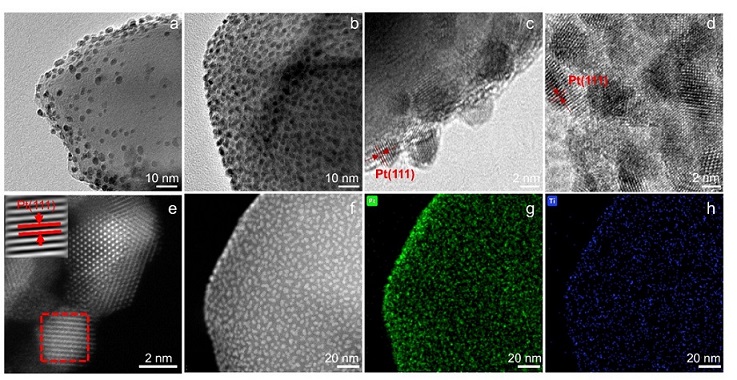
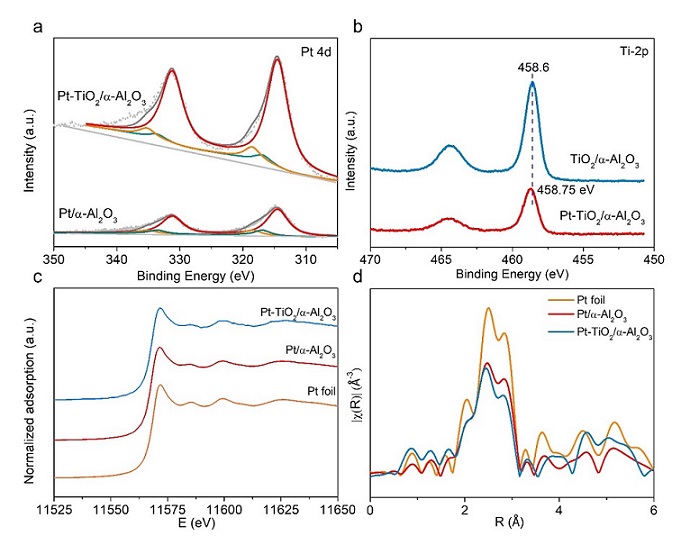
图2 催化剂电子结构表征
测试Pt-TiO2/α-Al2O3和 Pt/α-Al2O3两种催化剂LA加氢性能(图3)。与Pt/α-Al2O3催化剂相比,Pt-TiO2/α-Al2O3催化剂性能大为提高,TOF值从2487h-1提高到4943h-1。将本文的催化剂与其他文章对比,活性也达到较高值,与报道最好的Ru催化剂的结果相当。将 Pt-TiO2/α-Al2O3催化剂装入固定床中进行稳定性实验。在150 ºC时,催化剂可稳定运行500 h而无明显失活。进一步将温度升至190 ºC,LA转化率接近100%,选择性维持在98%左右,继续稳定运行500 h无明显失活。由此可见Pt-TiO2/α-Al2O3催化剂稳定性良好。
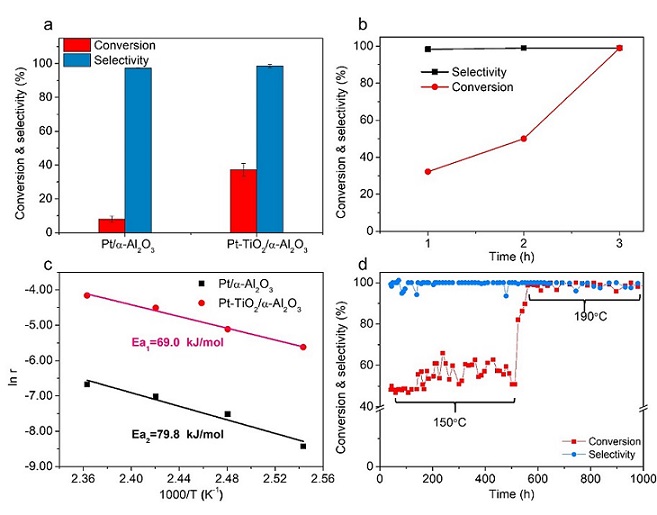
图3 催化剂乙酰丙酸水相加氢性能
利用CO- DRIFT 来区分Pt表面不同活性位点(图4a)。Pt-TiO2/α- Al2O3上CO线性吸附可拟合为三个峰,分别位于2087 cm-1、2063 cm-1和2044 cm-1,分别表示为α、β和γ。而Pt/α-Al2O3上仅观察到β和γ位。由于Pt/α- Al2O3和Pt-TiO2/α- Al2O3 中Pt纳米粒子具有相同的粒径和晶格结构,我们可以推断α位点源于Pt- TiO2相互作用。为了探索上述位点对LA吸附的作用,我们在原位LA表面吸附后进行了CO-DRIFTS(图4b, 4c)。LA在Pt/α-Al2O3表面饱和吸附后,β位的CO吸附峰减小,而γ位的CO吸附峰保留。LA占据了部分β位,而γ位不能吸附LA。Pt-TiO2/α- Al2O3催化剂上吸附LA后,除β位被占据外,α位消失。因此,Pt-TiO2相互作用产生的Pt物种能促进 LA在催化剂表面的富集。此外XPSC1s结果检测到反应后Pt-TiO2/α-Al2O3上产生COOH物种,进一步说明示LA在催化剂表面的富集(图4d)。
我们在LA饱和吸附后进行CO-DRIFTS反应,并在150℃常压下在H2中进一步反应,以揭示LA在不同位点上的反应活性。对于Pt/α-Al2O3,由于LA和中间体的吸附,β位峰面积降低,而γ位的数量没有变化。Pt-TiO2/α-Al2O3的β位全部消失,γ位点数量略有降低。因此,Pt的α位点上吸附LA用于进一步加氢产生的中间体进一步吸附在Pt纳米颗粒的β位点,而γ位点对LA和加氢中间体吸附较弱。不同表面Pt位点的电子密度按α位<β位<γ位的顺序递增,界面上的Pt缺电子α位有利于LA的强吸附,而β位Pt是中间体吸附位点。
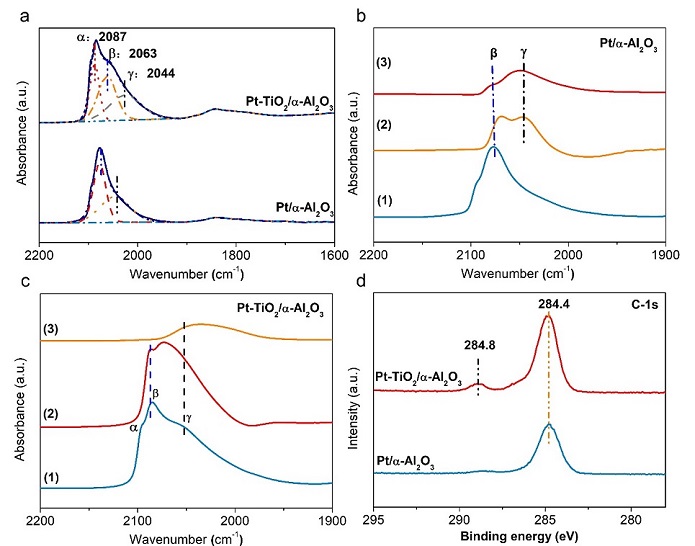
图4 LA吸附和加氢的表面Pt位点
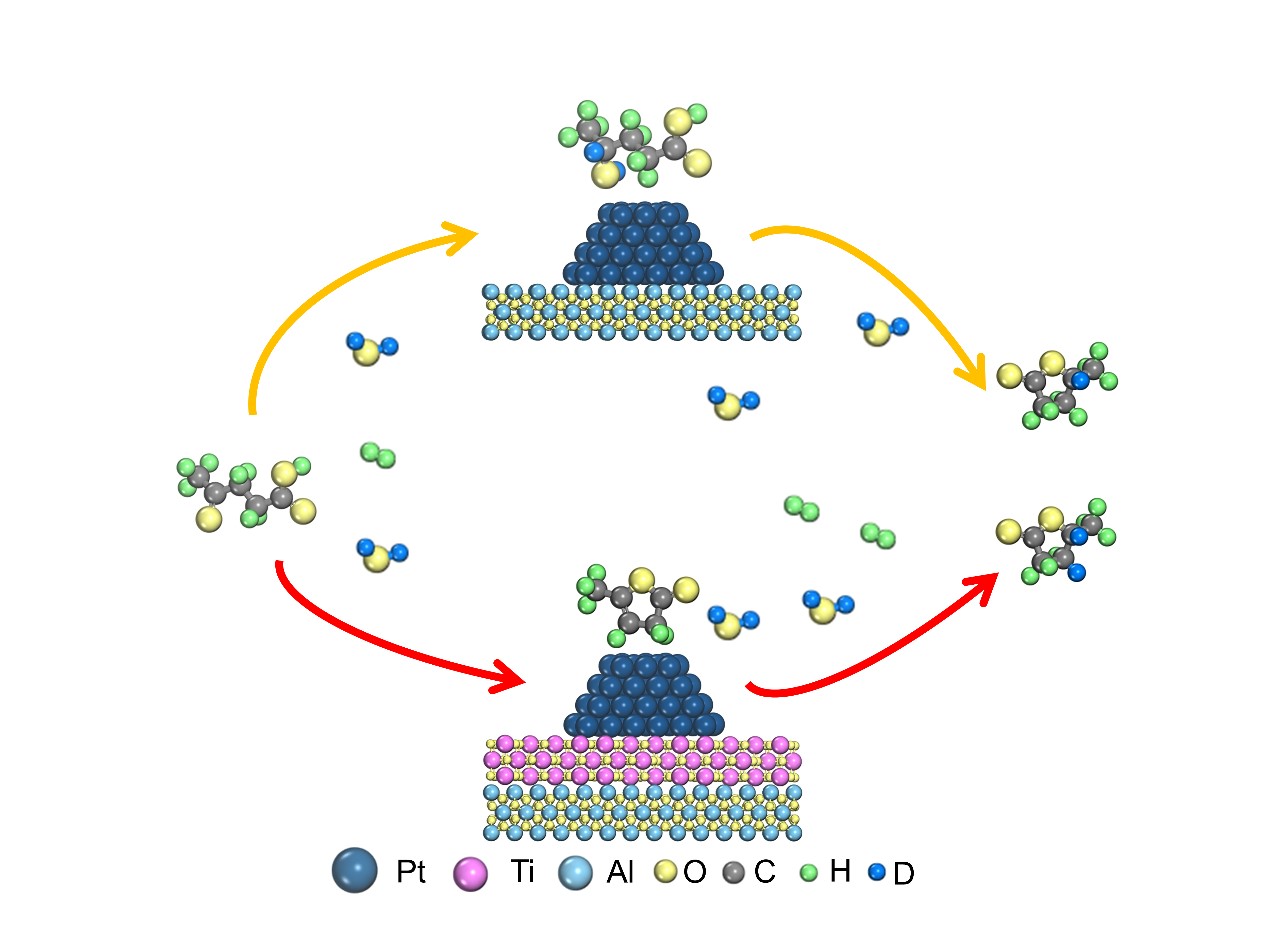
利用同位素实验研究了LA加氢反应机理。由于D2对LA加氢生成GVL几乎没有同位素效应(kH2/kD2≈1,图5a),因此氢气的活化不是LA水相加氢的限速步骤。用GC-MS对产物进行分析(图5d)。当D2为反应物时,GVL中未观察到D原子。也就是说,产物GVL中的H主要来自H2O溶剂。然后,将H2O替换为D2O,测试Pt/α-Al2O3和Pt-TiO2/α-Al2O3LA加氢反应性能。有趣的是,当以D2O中为溶剂,两种催化剂的催化活性都增强,检测到显著的H2O/D2O逆动力学同位素效应,其中Pt/α-Al2O3的KIE值为0.78(图5b),Pt-TiO2 /α-Al2O3的KIE值为0.52(图5c)。因此,水间接参与了反应,在Pt-TiO2相互作用形成后,水的作用更为显著。
分析Pt/α-Al2O3(图5e)和Pt-TiO2/α-Al2O3(图5f)在D2O溶剂中的产物可进一步了解反应路径。产物GVL上没有D原子时,其m/z为100。[1D]-GVL(GVL上有一个D原子)m/z为101,是Pt/α-Al2O3为催化剂的主要产物,[2D]-GVL(GVL上有两个D原子)m/z为102,是Pt-TiO2 /α-Al2O3催化剂的主要产物。这表明,Pt-TiO2相互作用形成后,水相LA加氢反应路线发生了改变。一般LA的加氢有两条路线(图6)。在路径1中,重水的D原子参与了LA的C=O键的加氢,进一步脱水环化生成[1D]-GVL。在路径2中,LA在酸性位点环化生成中间产物α-当归内酯,然后再加氢生成[2D]-GVL。因为Pt-TiO2/α-Al2O3上[2D]-GVL是主要产物,所以Pt-TiO2相互作用的存在使得LA加氢反应的路线由1路线转变为2路线。
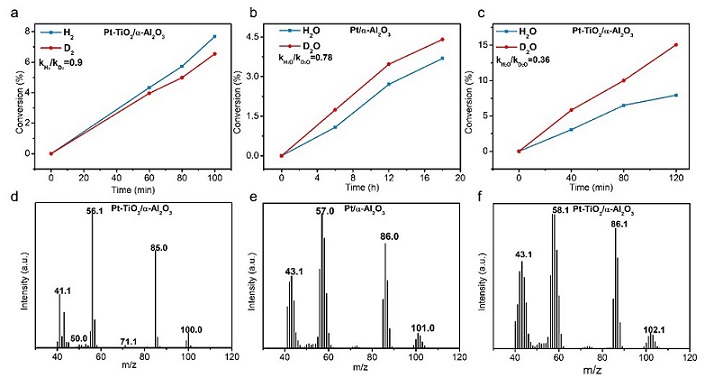
图5 动力学同位素实验
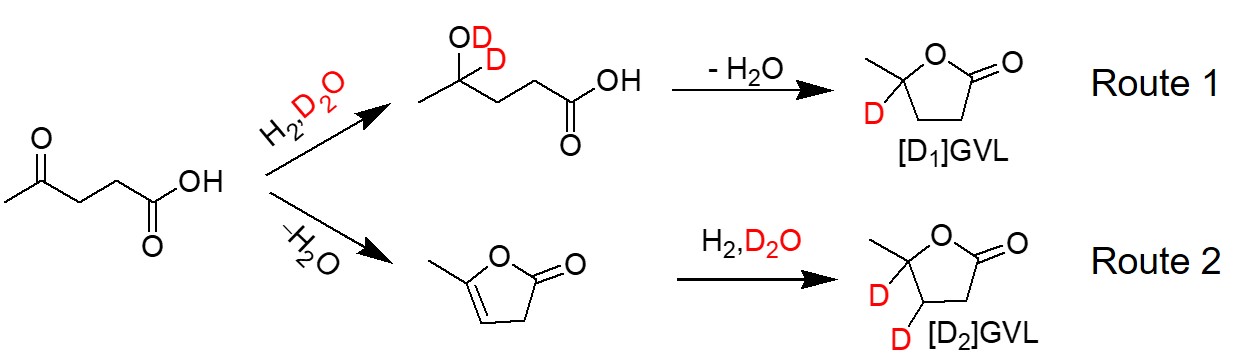
图6 LA加氢生成GVL的反应路径
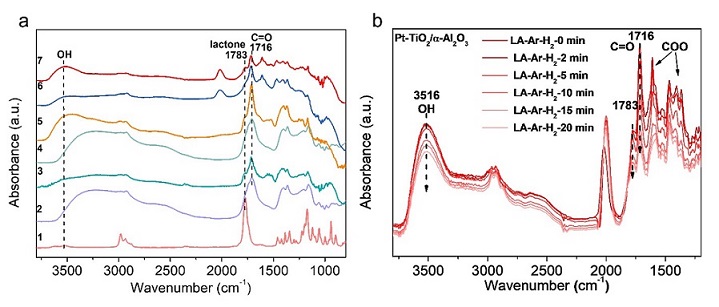
图7 LA水相加氢中间体的探究
原位FTIR测量可进一步揭示表面反应机理。图7a为150℃条件下 Ar气氛下Pt/α-Al2O3、Pt-TiO2/α-Al2O3和TiO2/α-Al2O3吸附LA的原位FTIR谱图。LA的羰基特征峰位于1716 cm-1。1783 cm-1处的峰是内酯的C=O的伸缩振动峰。注入LA水溶液到催化剂表面,α-Al2O3和Pt/α-Al2O3上只检测到LA的特征峰;与之不同,TiO2/α-Al2O3和Pt-TiO2/α-Al2O3上则检测到经LA环化产物中间体α-当归内酯在1783 cm-1处新的C=O伸缩振动峰。该结果与同位素实验结果一致,表明Pt-TiO2相互作用的形成改变了反应路径。我们还进行了水和LA吸附的对照实验,以揭示水在反应路径转换中的作用。当在TiO2/α-Al2O3上引入纯LA时,只观察到LA的峰;进一步注入水后可在1783 cm-1处发现生成了新的内酯吸收峰,且发现了TiO2 (Ti-OH)上的孤立羟基在3516 cm-1处的振动峰。由此可见,水的存在导致催化剂表面产生新的Ti-OH物种,为LA直接环化提供酸性位点。此外,由于Pt-TiO2/α-Al2O3表面Ti-OH和内酯峰强度更高,Pt-TiO2相互作用增强了对水的吸附和Ti-OH的形成。
为了进一步证实内酯是中间体,我们进行了原位红外反应。当在α-Al2O3和TiO2/α-Al2O3吸附LA的样品表面引入10%H2/Ar时,红外光谱没有变化,说明两个样品是没有LA加氢活性的。相反,Pt-TiO2/α-Al2O3 和Pt/α-Al2O3 的红外光谱显示,当通入10%H2/Ar后,吸光度逐步降低,这是因为发生了LA加氢反应(图7b)。与Pt/α-Al2O3相比,Pt-TiO2/α-Al2O3 的LA吸收峰降低更显著,表明其加氢效率更高。对于Pt/α-Al2O3, 通入H2后只消耗LA。而Pt-TiO2/α-Al2O3除消耗LA外,还消耗α-当归内酯和Ti-OH物种。因此,α-当归内酯是LA水相加氢的主要中间体,是Pt-TiO2相互作用的结果。Ti-OH物种在反应过程中不断动态消耗和生成。
综上所述,本工作通过直接排除Pt粒径、晶面结构、载体形态、孔结构和电子状态的影响,揭示了Pt-TiO2相互作用诱导LA水相加氢反应路径变化提升反应效率的新机制。研究表明,在水和氢的作用下,Pt-TiO2相互作用导致更多Ti-OH活性位点的生成,促进了LA的吸附和直接脱水环化成α-当归内酯中间体,中间体之后传递到Pt平面β位点并与活性氢反应生成产物γ-戊内酯。反应路径由常见的直接加氢再环化转变为直接环化再加氢过程。这一工作为水相加氢高效催化剂的设计和揭示金属催化剂水相中的表界面多位点协同机制提供了新思路。
原文链接:
https://www.sciencedirect.com/science/article/pii/S0926337322011778

 当前位置:
当前位置: