金属亚纳米催化剂的电子和几何结构显著依赖于其包含的金属原子个数(核数),并最终决定其催化性能,即所谓的核数效应(nuclearity effects)。探明核数效应的本质对于理性设计合成高效、高金属利用率的亚纳米催化剂具有重要意义,但仍是一项重大挑战。有鉴于此,中科院山西煤化所陈朝秋/覃勇研究团队在前期对原子层沉积过程(ALD)中金属成核与生长规律的研究基础上(ACS Catal. 2021, 11, 7, 4146–4156;ACS Catal. 2020, 10, 4, 2837–2844),进一步利用载体表面配位环境耦合ALD逐层生长特点精准合成了Fe亚纳米金属催化剂(从单原子、双原子、三原子到四原子团簇)。 Fe亚纳米催化剂的电子结构和自旋构型表现出显著的核数依赖性,并影响H2O2的活化行为和所生成活性氧物种的氧化能力,最终决定其氧化苯分子活性。 其中,Fe单原子催化剂由于具有独特的配位结构(Fe1N2O3)和中自旋态(t2g4eg1),展现出超高的苯选择性氧化制苯酚活性,分别是Fe双原子、三原子和四原子团簇催化剂的3.4、5.7和13.6倍。本工作可为亚纳米催化剂和活性中心结构的精确设计和深入认识自旋及核数效应等亚纳米催化规律提供指导和借鉴。
背景介绍
将金属催化剂的尺寸降低到亚纳米尺度(单原子和团簇)可获得独特的物理化学性质和极高的原子利用率,有望突破传统催化剂的限制,获得更高的催化效率和选择性。在亚纳米尺度,金属催化剂核数的极小变化(比如增加或减少一个金属原子)会对其本征电子性质和几何结构产生重要影响,进而显著改变其催化性能,即所谓的核数效应。因此,深入研究并理解核数效应的本质对于理性设计高性能金属亚纳米催化剂具有重要意义,但仍充满挑战。主要原因之一是目前仍缺乏有效的精准合成具有可控原子数的亚纳米催化剂的方法。ALD技术是一种可精确构建具有可控原子数亚纳米催化剂的理想方法。然而,目前利用ALD制备原子数可控(单原子或团簇)的亚纳米催化剂的成功案例非常有限,这主要是由于制备过程中表面吸附分子的移动、金属原子成核与生长竞争等原因导致。
与传统金属催化剂的连续能带结构不同,亚纳米催化剂具有离散的,类分子的能级结构。这就决定了亚纳米催化剂中d轨道电子性质将在催化过程中起着更重要的作用,特别是催化剂的电子自旋构型,已经被认为是一个关键的催化性能描述符。但是关于亚纳米催化剂核数-自旋构型-催化性能间相互关系的知识仍是空白。
苯酚作为一种重要的工业原材料,需求广泛,且需求量在不断增加。温和条件下利用过氧化氢将苯选择性催化氧化为苯酚被认为是一种高效环保的苯酚生产工艺。据文献报道,Fe,Cu,Co等单原子催化剂展现出显著优于相应纳米颗粒催化剂的催化苯氧化制苯酚性能。然而,为了满足工业化生产需求,苯氧化性能仍需进一步提高。另一方面,该反应是典型的结构敏感反应,因此急需系统研究核数效应对亚纳米催化剂催化苯氧化制苯酚性能的影响规律,帮助开发更高效的亚纳米催化剂。
本文亮点
1、 以N,O共掺杂碳纳米棒上丰富的N,O杂原子为锚定位,利用ALD精确调控Fe的生长,可控合成了核数可调的Fe基亚纳米催化剂(Fex-NOC, x代表Fe原子个数,x=1-4);
2、 Fex-NOC的电子性质和几何结构以及自旋构型显著依赖于其核数。随着核数增加,Fe的价态逐渐降低,金属性增强,自旋逐渐降低(中自旋到低自旋)。
3、 Fex-NOC表现出优异的催化苯选择性氧化制苯酚活性,其活性随核数增加而大幅降低。其中Fe1-NOC单原子催化剂活性最强,60 oC下TOF高达1869 h-,分别是Fe2-NOC、Fe3-NOC和Fe4-NOC催化剂的3.4、5.7和13.6倍。此外,25℃时Fe1-NOC催化剂的TOF为407 h-,远高于目前文献报道值。
4、 实验和理论计算结果表明Fex-NOC催化剂的电子结构和自旋构型随核数的增加而改变,进而影响H2O2的活化行为和所生成活性氧物种的氧化能力,最终决定其氧化苯分子活性。Fe1-NOC的优异性能可归功于其独特的配位结构(Fe1N2O3)和自旋构型(t2g4eg1)。中自旋Fe1-NOC催化剂展现出较弱的活性O物种吸附强度,这有利于苯酚中C-O键的形成,进而展现出高苯氧化制苯酚活性。

图1. Fex-NOC催化剂制备流程、球差电镜图片及EELS谱图。首先在氩气氛围下热解聚苯胺纳米棒,得到N,O共掺杂碳载体(NOC)。随后利用ALD在NOC上沉积Fe物种,得到Fex-NOC催化剂。从球差电镜(AC–HADDF–STEM)图片可知,当ALD循环次数为1,5,10,和20时,分别得到以Fe单原子(Fe1-NOC)、双原子(Fe2-NOC)、三原子(Fe3-NOC)、和四原子(Fe4-NOC)团簇为主的催化剂。原子分辨的EELS表明Fe物种与N,O配位。
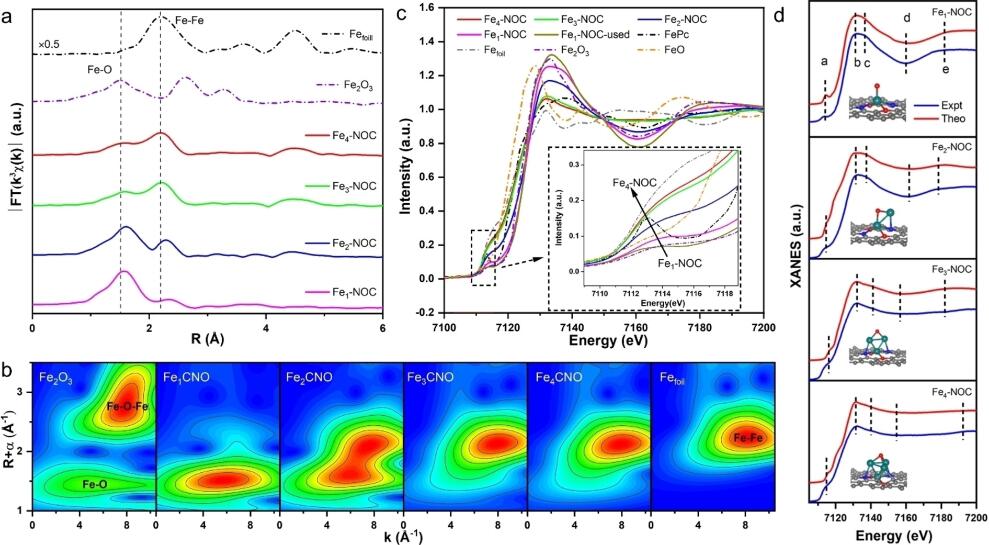
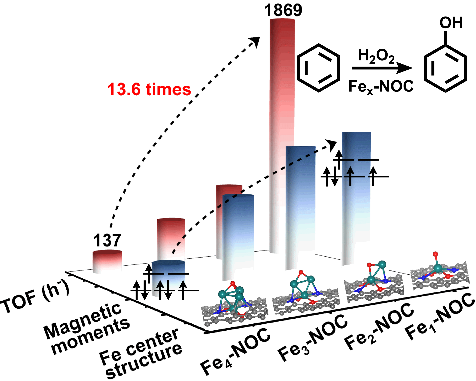
图2. 不同催化剂中Fe位点结构确认。通过X射线吸收精细谱对不同样品中Fe单原子及Fe2-Fe4团簇的电子性质和几何结构进行分析。由图2a可知,Fe1-NOC中只存在Fe-N(O)配位,表明Fe物种是原子级分散的。与之相比,Fe2–NOC、Fe3–NOC和Fe4–NOC中除Fe-N(O)配位外,还存在明显的Fe-Fe配位(图2b,小波变换支持上述结论),且随着Fe ALD循环次数的增加,Fe-Fe配位逐渐增强,Fe物种价态逐渐降低(图2c)。对EXAFS数据拟合可知,Fe1-NOC中Fe原子分别与两个N原子和三个O原子配位,无Fe-Fe配位,而Fe2–NOC、Fe3–NOC和Fe4–NOC中Fe-Fe配位分别为1.1、1.9和2.9,表明形成了Fe2、Fe3和Fe4团簇,这与球差电镜结果一致。基于上述AC–HADDF–STEM 、EELS和EXAFS拟合结果,我们利用DFT模拟确定了Fe1–NOC、Fe2–NOC、Fe3–NOC和Fe4–NOC中Fe物种活性位结构分别为Fe1N2O3、Fe2N2O3、Fe3N2O3和Fe4N2O3C2。基于这些结构的XANES模拟谱也与实际测试谱线一致(图2d),进一步证实上述活性位结构的合理性。
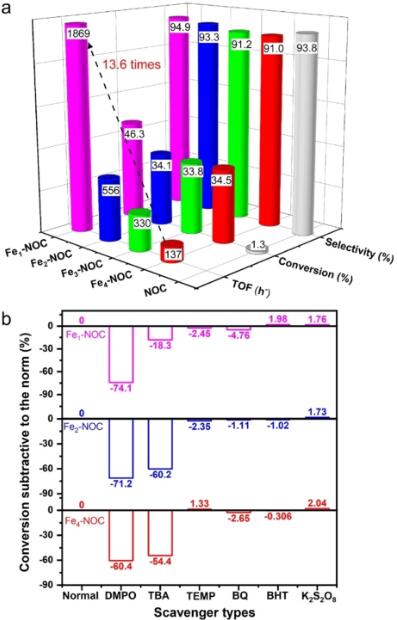
图3. 不同催化剂的苯氧化性能及反应过程中H2O2活化方式。将Fex–NOC催化剂用于常压下苯氧化反应。结果表明,Fex–NOC催化苯氧化活性表现出显著的核数效应,Fe单原子催化剂展现出最高的苯氧化活性(高达1869 h-1)。随着核数增加,催化剂催化苯氧化活性大幅降低。
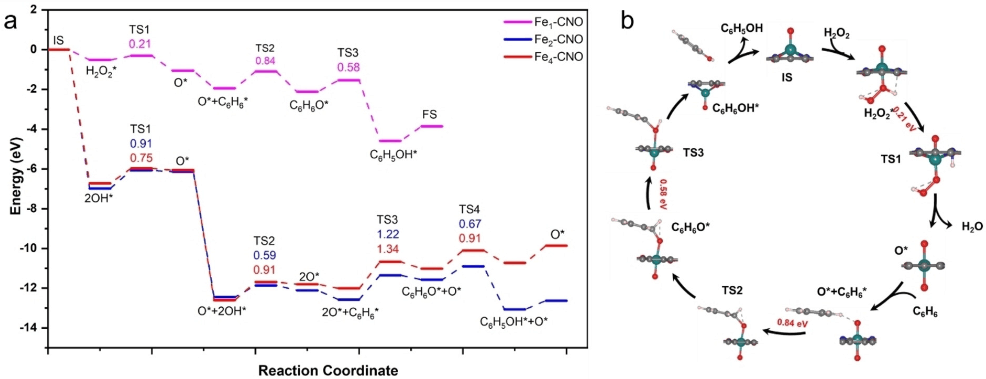
图4. DFT模拟不同催化剂苯氧化反应路径。为了更深入的理解苯氧化反应中Fex-NOC催化剂的核数效应,我们进行了自由基捕获实验(图3b)、同位素实验以及反应过程DFT模拟分析。结果表明,H2O2首先在Fe单原子位点活化生成*OOH和*H,随后*OOH和*H迅速生成一个水分子及吸附在Fe位点的活性O。紧接着,活性O原子与苯中C原子结合形成C-O键,该过程为反应的决速步。最后,经过H转移过程以及苯酚脱附过程,活性位点得以再生(动力学同位素实验排除了芬顿反应的发生)。与之相比,Fe2-NOC和Fe4-NOC为代表的Fe团簇催化剂,需要两个H2O2在Fe位点通过生成*OH中间体活化为两个活性O,且在随后的C-O键形成过程中,活化能垒高达1.22和1.34 eV。因此,由于较低的活性O物种和C-O形成能垒,Fe1-NOC展现出最高的苯氧化活性。

图5. 不同催化剂间活性差异原因分析。根据文献报道可知,活性O物种在金属位点的吸附强度直接影响苯酚中C-O键形成,因此,进一步分析了活性O物种在Fe位点的吸附强度以阐明不同催化剂决速步能垒差异的根本原因。由图5a-f可知,随着Fe原子数增加,Fe的d轨道与活性O原子的p轨道相互作用逐渐增强(图5g,O2-TPD证明了这一点),而过强的Fe-O相互作用不利于苯酚中C-O键的形成。为了进一步揭示活性O原子在Fex-NOC上吸附差异的根本原因,我们计算并测量了不同催化剂磁性性质。结果表明随着Fe原子数增加,Fe位点由中自旋逐渐变为低自旋,自旋态的降低增强了活性O物种的吸附,使其氧化能力减弱。综上,随催化剂中Fe原子数降低,Fe位点由低自旋变为中自旋,使得活性O物种在Fe位点的吸附强度减弱,进而有利于苯酚中C-O键形成(降低了决速步能垒)。
总结与展望
综上所述,本工作结合N,O杂原子的强配位能力和ALD原子级控制精度的优势,成功实现了N,O共配位的Fe亚纳米催化剂的精准合成,为原子尺度下系统研究核数-性能关系提供了平台。改变团簇中Fe原子个数可以调节其电子性质和Fe活性位点的自旋构型,从而调控Fe位点上H2O2的活化方式和活性O的吸附强度,使其更有利于C-H氧化。这项工作为理解亚纳米催化剂中自旋构型控制的核数效应及其对催化性能的影响规律提供了深刻的见解,并为合理设计高效的低核数金属亚纳米催化剂开辟了道路。
文章链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202216062

 当前位置:
当前位置: